Antibiotics-β-lactam antibiotics(penicillins,cephalosporins ),tetracycline,macrolides,aminoglycoside antibiotics
Free pharmacy material































Antibiotics
Antibiotics are substances produced by various species of microorganisms (bacteria, fungi, actinomycetes) that suppress the growth of other microorganisms and eventually may destroy them. Since their discovery in the 1930s, antibiotics have made it possible to cure diseases caused by bacteria such as pneumonia, tuberculosis, and meningitis – saving the lives of millions of people around the world.
Historically, the most common classification has been based on chemical structure and proposed mechanism of action, as follows:
- Agents that inhibit the synthesis of bacterial cell walls; these include the penicillins and cephalosporins, which are structurally similar, and dissimilar agents such as cycloserine, vancomycin, bacitracin, and the imidazole antifungal agents
- Agents that act directly on the cell membrane of the microorganisms affecting permeability and leading to leakage of intracellular compounds; these include polymyxin, polyene antifungal agents, nystatin and amphotericin B, which bind to cell wall sterols
- Agents that affect the function of 30S and 50S ribosomal subunits to cause reversible inhibition of protein synthesis; these include tetracyclines, erythromycins, chloramphenicol, and clindamycin
- Agents that bind to the 30S ribosomal subunit and alter protein synthesis, which eventually leads to cell death; these include aminoglycosides
- Agents that affect nucleic acid metabolism such as rifamycins, which inhibit DNA-dependent RNA polymerase
26.1 β-LACTAM ANTIBIOTICS
Antibiotics that contain the β-lactam (a four-membered cyclic amide) ring structure constitute the dominant class of agents currently employed for the chemotherapy of bacterial infections. Penicillin, cephalosporin, and their semi-synthetic derivatives come under this class.
Mechanism of action: The cell wall of bacteria is essential for the normal growth and development. Peptidoglycan is a heteropolymeric component of the cell wall that provides rigid mechanical stability by virtue of its highly cross-linked lattice-wise structure.
The peptidoglycan is composed of glycan chains, which are linear strands of two alternating amino sugars (N-acetyl glucosamine and N-acetyl muramic acid) that are cross-linked by peptide chains.
The biosynthesis of peptidoglycan involves three stages.
The last step in peptidoglycan synthesis is inhibited by β-lactam antibiotics. The transpeptidase enzyme contains serine, which is probably acylated by β-lactam antibiotics with cleavage of -CO-N- bond of the β-lactam ring. This renders the enzyme inoperative and inhibits peptidoglycan synthesis.
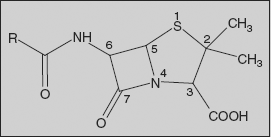
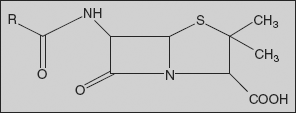
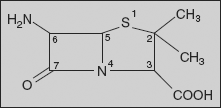
Penicillins: The penicillins are commonly named as penams, a designation in which the sulphur atom is given the top priority.
Using this nomenclature, the penicillins have a prerequisite carboxylic acid group placed at the C-3 position. The west-end substituents are joined to the C-6 centre and are usually substituted via acylation, thus constituting a variety of C-6 acylamido substituents. The β-lactam carbonyl centre is located at position 7, and the C-2 centre contains a geminal dimethyl substitution characteristic of penicillins.
Classification
1. Early penicillins
General impact on antibacterial activity:
- Excellent gram-positive potency against susceptible Staphylococci, Streptococci
- Useful against some gram-positive cocci
- Good oral absorption but relatively acid-labile
- Ineffective against gram-negative bacilli
- Susceptible by deactivation by penicillinase
2. Penicillinase-resistant penicillins
General impact on antibacterial activity:
- Diminished susceptibility to many penicillinases; active against microorganisms resistant to early penicillins
- Oxacillins offer good oral activity
- Inadequate spectrum against many gram-negative species
3. Broad-spectrum penicillins
- Extended spectrum of activity against some gram-negative bacteria; retention of gram-positive potency
- Generally well-absorbed orally; ampicillin can be dosed I.V. and I.M. as well; amoxycillin exceptional oral agent
- Prodrug esters (of ampicillin) enhance systemic drug levels
- Ineffective against Pseudomonas aeruginosa
4. Anti-pseudomonal penicillins
- Extended spectrum of activity against many pathogenic gram-negative bacteria; reduced gram-positive potency
- Good activity against P. aeruginosa
- Oral absorption, chemical stability problematic
- Prodrug esters (of carbenicillin) enhance systemic drug levels
5. Broad-spectrum ureido penicillins
- Enhanced spectrum of activity against P. aeruginosa, expanded activity against Klebsiella, Serratia, Proteus
- Good potency against gram-positive bacteria, but generally not effective against penicillinase producers
- Good pharmacokinetic profile
6. Penicillin with a C-6 amidino west-end
- Good activity against E. coli, Klebsiella, Shigella, Salmonella, and many other resistant species
- Inactive against P. aeruginosa
- Prodrug esters enhance systemic drug levels
Mechanism: Refer to β-lactam antibiotics
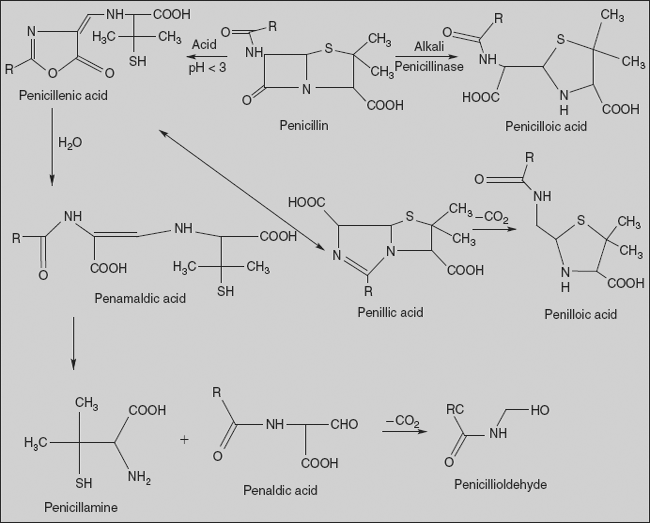
Chemical degradation of penicillins
In strongly acidic solutions (pH<3), penicillin undergoes a complex series of reactions leading to a variety of inactive degradation products. Similarly, penicillinase enzyme hydrolyses the β-lactam ring to produce inactive penicilloic acid.
Acid-catalysed degradation in stomach contributes in a major way to the poor oral absorption of penicillin. Thus, efforts to obtain penicillins with improved pharmacokinetic and microbiologic properties have sought to find acyl functionalities that would minimize sensitivity of the β-lactam ring to acid hydrolysis and, at the same time, maintain antibacterial activity.
Substitution of an electron-withdrawing group in the α-position of benzyl penicillin has stabilized the penicillin to acid-catalysed hydrolysis. The increased stability imparted by such electron-withdrawing groups has been attributed to a decrease in reactivity of the side-chain amide carbonyl oxygen atom toward participation in β-lactam ring opening to form the penicillenic acid.
Adverse effects Penicillins cause hypersensitivity reactions (allergy) in several percentages of patients and this may be due to formation of antigenic penicilloyl proteins formed in vivo by the reaction of nucleophilic groups (e.g., ε-amino) on specific body proteins with the β-lactam carbonyl groups.
The first mode of β-lactam resistance is due to enzymatic hydrolysis of the β-lactam ring. If the bacterium produces the enzymes β-lactamase or penicillinase, these enzymes will break open the β-lactam ring of the antibiotic, rendering the antibiotic ineffective. The second mode of β-lactam resistance is due to possession of altered penicillin-binding proteins (PBP). β-lactams cannot bind as effectively to these altered PBPs, and, as a result, the β-lactams are less effective at disrupting cell-wall
26.1.1 Sar of Penicillins
- C-6 Amino west-end substitution:
- The design and development of the west-end substituents has been aimed at strengthening various weaknesses that have traditionally hampered penicillins in terms of activity, stability, resistance, and absorption/distribution.
- The C-6 amine moiety itself is necessary for appreciable antibacterial activity, but substitution of the amine via monoacylation can offer much more potent congeners.
- Only carboxamido-derived west-end moieties are tolerated; sulphonation- or phosphoramide-containing substituents are devoid of antibacterial activity. Similarly, imide- or carbamate-containing west-end are inferior.
- Agents that were stable to penicillinase enzymes were created by introducing a more crowded environment around β-lactam moiety. Methicillin contains 2, 6-dimethoxy benzamido west-end and the placement of methoxy groups on the aromatic ring is important; the bis ortho arrangement creates the most effective crowding around the β-lactam carbonyl centre, while retaining good activity.
- The oxacillins contain 5-methyl-3-phenyl-4-isoxazolyl west-end substituents that similarly impose crowding in proximity to the β-lactam ring. In these compounds, both the methyl and phenyl substituents are positioned closest to β-lactam system. Removal of either group increases susceptibility to penicillinases.
- To expand the antibacterial spectrum of penicillins, more hydrophilic west-end was designed that can enhance potency against gram-negative pathogens. Ampicillin contains a D-α-aminophenylacetamido west-end and is most recognized as amino penicillins. In general, substituents on the phenyl ring are detrimental either due to decreased hydrophilicity or, conversely, due to adverse polar effects if an ionizable substituent is present. A notable balance of these opposing forces has been achieved with the placement of a para-hydroxyl group on to the phenyl ring. Amoxycillin is essentially comparable to ampicillin in terms of in vitro potency, but displaces better oral efficacy.
- The spectrum of activity was further expanded with introducing strongly acidic groups at the α-carbonyl centre of the side-chain. These imparted useful potency against P. aeruginosa. Carbenicillin possesses α-carboxy phenyl acetamido west-end.
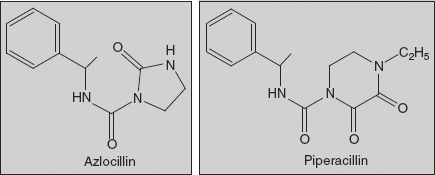
- The acylation of the ampicillin west-end amine functionality with certain polar groups leads to cyclic urea derivatives, ureido penicillins (azlocillin), which contain a five-membered cyclic urea system joined via N-acylation to the α-amino substituent of ampicillin. These are more active against P. aeruginosa than carbenicillin and possess potency against other gram-negative pathogenic species. The presence of urea group imparts improved penetration into these gram-negative species traditionally resistant to penicillins.
- Substituents at sulphur:
- Sulphur is the only atom at position 1 of the penicillin in order to retain appreciable antibacterial activity.
- C-2 substituents:
- The geminal dimethyl group at C-2 is characteristic of the penicillin.
- C-3 substituents:
- In general, derivatization of the C-3 carboxylic acid functionality is not tolerated unless the free penicillin carboxylic acid can be generated Antibiotics. Doubly activated penicillin esters such as alkanoyloxyalkyl congeners undergo rapid cleavage Antibiotics to generate active penicillin—e.g., pivampicillin and becampicillin.
- Variation at N-4:
- The nitrogen atom at the ring junction is vital for antibacterial activity; the nitrogen atom contributes to the reactivity of the β-lactam carbonyl centre.
Methicillin [2S-(2α,5α,6β)]-3,3-Dimethyl-7-oxo-6-(2,6-dimethoxybenzamido)-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
Synthesis
It is synthesised by acylating 6-APA with 2,6-dimethoxybenzoic acid in the presence of thionyl chloride .
Oxacillin [2S-(2α,5α,6β)]-3,3-Dimethyl-7-oxo-6-(5-methyl-3-phenyl-4-isoxazolcarboxamido)-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
Synthesis
Oxacillin is synthesised by reacting 5-methyl-3-phenyl-4-isoxazolcarboxylic acid chloride with 6-APA in the presence of sodium bicarbonate. The 5-methyl-3-phenyl-4-isoxazolcarboxylic acid chloride is synthesised by the following method. Reacting benzaldehyde with hydroxylamine gives oxime, which when oxidized by chlorine gives benzhydroxamic acid chloride. This is reacted with methylacetoacetate in the presence of sodium methoxide, giving the methyl ester of 5-methyl-3-phenyl-4-isoxazolcarboxylic acid. Alkaline hydrolysis of the resulting ester gives the corresponding acid, which is reacted with thionyl chloride to give the acid chloride necessary for acylation.
Cloxacillin [2S-(2α,5α,6β)]-3,3-Dimethyl-7-oxo-6-[(5-methyl-3-(o-chlorphenyl)-4-isoxazol-carboxamido)]-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
It is synthesised from o-chlorobenzaldehyde by the scheme described above.
Synthesis
Dicloxacillin [2S-(2α,5α,6β)]-3,3-Dimethyl-7-oxo-6-[(5-methyl-3-(2,6-dichlorophenyl)-4-isoxazol-carboxamido)]-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid.
It is also synthesised by the scheme described above, using 2,6-dichlorobenzaldehyde as the starting substance.
Ampicillin [2S-[2α,5α,6β(S)]]-3,3-Dimethyl-7-oxo-6-(2-amino-2-phenylacetamido)-4-thia-azabicyclo[3.2.0]-heptan-2-carboxylic acid
Synthesis
Acylation of 6-APA with the 2-azidophenylacetyl chloride gives the corresponding amide; catalytic reduction of azido group affords the amine ampicillin.
The corresponding product from acylation with 2-azido-4-hydroxyphenyl acetyl chloride isamoxycillin.
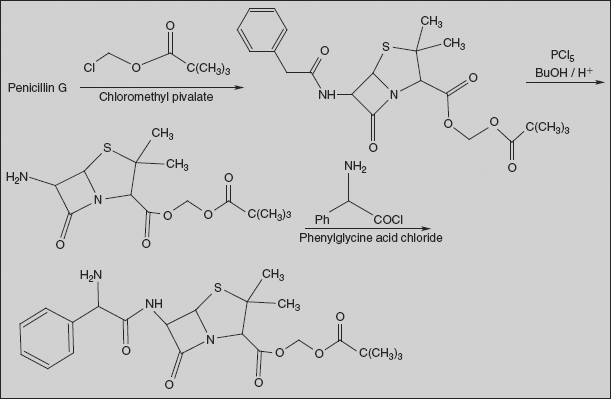
Pivampicillin 2,2-Dimethylpropanoyloxymethyl (2S,5R,6R)-6-{[(2R)-2-amino-2-phenylacetyl]amino}-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylate
Pivampicillin is a pivaloyloxymethylester of ampicillin. It is a prodrug that is thought to enhance the oral bioavailability of ampicillin because of its greater lipophilicity compared to that of ampicillin.
Synthesis
Benzyl penicillin is esterified with chloromethyl pivalate; acid hydrolysis of amide bond gives 6-APA ester derivative. Acylation of this with phenylglycine acid chloride affords pivampicillin.
Carbenicillin [2S-(2α,5α,6β)]-3,3-Dimethyl-7-oxo-6-(2-carboxy-2-phenylacetamido)-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
Synthesis
Carbenicillin is synthesised by direct acylation of 6-APA in the presence of sodium bicarbonate by phenylmalonic acid monobenzyl ester chloride, which forms the benzyl ester of carbenicillin, the hydrogenolysis of which using palladium on carbon or calcium carbonate as catalyst gives the desired product.
Ticarcillin [2S-(2α,5α,6β)]-3,3-Dimethyl-7-oxo-6-[2-carboxy-2-(3-thienyl)acetamido]-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
It is also synthesised by direct acylation of 6-APA in the presence of sodium hydroxide, but with 3-thienylmalonic acid chloride, which gives ticarcillin.
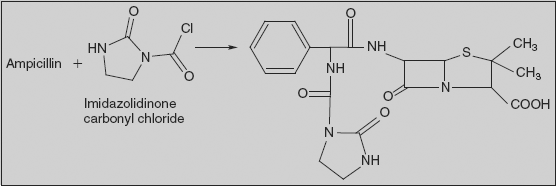
Azlocillin (2S,5R,6R)-3,3-Dimethyl-7-oxo-6-[(R)-2-(2-oxoimidazolidin-1-carboxamido)-2-phenylacetamido]-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
Synthesis
Azlocillin is synthesised by directly reacting 1-chlorocarbonyl-2-imidazolidinone (2-imidazolidinone is acylated with phosgene, forming 1-chlorocarbonyl-2-imidazolidinone) and ampicillin.
Synthesis
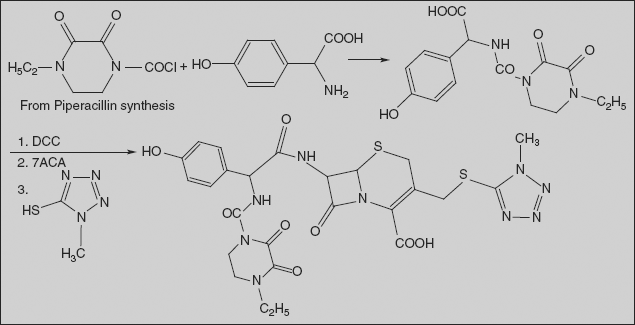
Piperacillin (2S,5R,6R)-3,3-Dimethyl-7-oxo-6-[(2R)-2-[(4-ethyl-2,3-dioxo-1-piperazinyl)formamido]-2-phenylacetamido]-4-thia-1-azabicyclo[3.2.0]-heptan-2-carboxylic acid
Synthesis
Piperacillin is also synthesised by acylating ampicillin, but with 1-chlorocarbonyl-4-ethylpiperazin-2,3-dione. The necessary 1-chlorocarbonyl-4-ethylpiperazin-2,3-dione is synthesised by reacting N-ethylethylenediamine with diethyloxalate, forming 4-ethylpiperazin-2,3-dione, and then acylating this with phosgene.
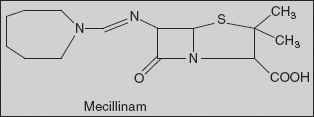
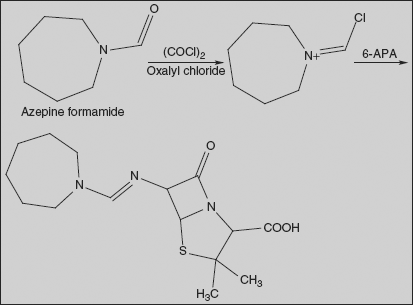
Mecillinam (Amdinocillin) (2S,5R,6R)-6-(azepan-1-ylmethylideneamino)-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
Reaction of azepine formamide with oxalyl chloride gives reactive derivative, which on condensation with 6-APA leads to the formation of the amidine mecillinam.
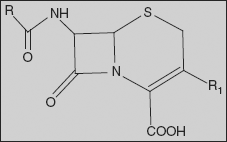
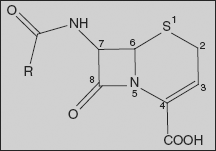
26.1.2 Cephalosporins
The cephalosporins are β-lactam antibiotics isolated from Cephalosporium species and/or prepared semi-synthetically. This comes under the class of 7-amino cephalosporonic acid (7-ACA) derivatives and are much more acid-stable than the corresponding 6-APA compounds. The cephalosporins have a mechanism of action similar to that of penicillins - mainly, they inhibit the cross-linking of the peptidoglycan units in bacterial cell wall by inhibiting transpeptidase enzyme.
The cephalosporin nucleus, 7-aminocephalosporanic acid (7-ACA), was derived from cephalosporin C and proved to be analogous to the penicillin nucleus 6-aminopenicillanic acid, but it was not sufficiently potent for clinical use. Modification of the 7-ACA side-chains resulted in the development of useful antibiotic agents, and the first agent cephalothin (cefalothin) was launched by Eli Lilly in 1964.
Classification
1. First-generation cephalosporins
These drugs have the highest activity against gram-positive bacteria and the lowest activity against gram-negative bacteria.
R
|
R1
| |
Cephalexin
|  |
CH3
|
Cefadroxil
|  |
CH3
|
Cephradine
|  |
CH3
|
Cephalothin
|  |
—CH2OCOCH3
|
Cephacetrile
|
—CH2OCOCH3
| |
Cefazolin
|  |  |
2. Second-generation cephalosporins
These drugs are more active against gram-negative bacteria and less active against gram-positive bacteria than first-generation members.
R
|
R1
| |
Cefaclor
|  |
Cl
|
Cefamandole
|  |  |
Cefuroxime
|  |
—CH2OCONH2
|
3. Third-generation cephalosporins
These drugs are less active than first-generation drugs against gram-positive bacteria, but have a much-expanded spectrum of activity against gram-negative organisms.
Degradation of cephalosporins Although cephalosporins are more stable to hydrolytic degradation reactions than penicillins, they experience a variety of chemical and enzymatic transformations, whose specific nature depends on the side-chain at C-7 and the substituent on C-3 atom.
The presence of a good leaving group at C-3 facilitates spontaneous expulsion of the 3’-substituent by concerted event due to hydrolysis of C-N bond of β-lactam nucleus by any general nucleophile or β-lactamase. Thus, desacetyl cefotoxime is more stable to hydrolysis in comparison to cefotoxime.
The absence of a leaving group at position 3 of cephalosporins makes them more acid-stable, thus rendering them suitable for oral consumption. Hence, cephalexin possessing methyl group at position 3 is much better absorbed than cephaloglycin having acetoxymethyl group at position 3, while both have identical phenylglycyl side-chain at the position 7. The nature of substituents at C-7 of cephalosporins plays an important role in determining the facility with which the reactive β-lactam bond is hydrolysed or broken either by chemical or enzymatic means of degradation of cephalosporin C.
1. In strong acid solution:
In the presence of esterase/acid, cephalosporin-C gave desacetyl cephalosporin and then inactive desacetyl cephalosporin lactone.
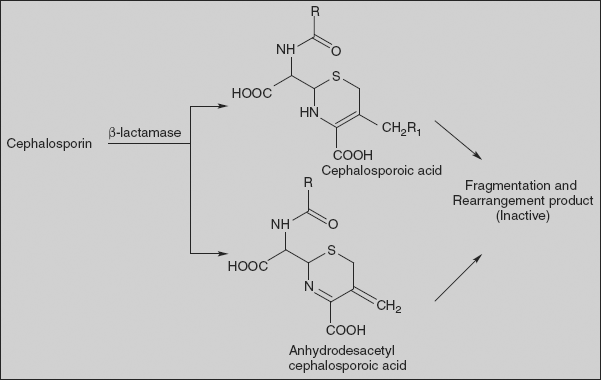
2. In the presence of β-lactamase:
The enzyme β-lactamase or cephalosporanase degraded cephalosporin C into cephalosporoic acid, anhydrodesacetyl cephalosporoic acid, and desacetyl cephalosporoic acid. Further breakdown of these acidic products leads to many other fragmented and rearranged products.
3. In the presence of acylase
On enzymatic degradation in the presence of acylase, cephalosporin C gives 7-amino cephalosporanic acid, which in the presence of acid undergoes lactonization to give inactive des- acetyl-7-amino cephalosporanic acid lactone.
SAR of cephalosporins
- 7-Acylamino substituents:
- Acylation of amino group generally increases the potency against gram-positive bacteria, but it is accompanied by a decrease in gram-negative potency.
- High antibacterial activity is observed only when the new acyl groups are derived from carboxylic acids for gram-positive bacteria.
- Substituents on the aromatic ring that increases lipophilicity provide higher gram-positive activity and generally lower gram-negative activity.
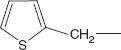
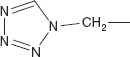
- The phenyl ring in the side-chain can be replaced with other heterocycles with improved spectrum of activity and pharmacokinetic properties, and these include thiophene, tetrazole, furan, pyridine, and aminothiazoles.
- C-3 substituents:
- The nature of C-3 substituents influences pharmacokinetic and pharmacological properties as well as antibacterial activity. Modification at C-3 position has been made to reduce the degradation (lactone of desacetyl cephalosporin) of cephalosporins.
- The benzoyl ester displaces improved gram-positive activity but lower gram-negative activity.
- Pyridine and imidazole-replaced acetoxy groups show improved activity against P. aeruginosa.Displacement of acetoxy group by azide ion yields derivatives with relatively low gram-negative activity.
- Displacement with aromatic thiols of 3-acetoxy group results in an enhancement of activity against gram-negative bacteria with improved pharmacokinetic properties.
- Replacement of acetoxy group at C-3 position with —CH3, Cl has resulted in orally active compounds.
- Cephamycins: Introduction of C-7 α-methoxy group shows higher resistance to hydrolysis by β-lactamases.
- Oxidation of ring sulphur to sulphoxide or sulphone greatly diminishes or destroys the antibacterial activity.
- Replacement of sulphur with oxygen leads to oxacepam (latamoxef) with increased antibacterial activity, because of its enhanced acylating power. Similarly, replacement of sulphur with methylene group (loracarbef) has greater chemical stability and a longer half-life.
- The carboxyl group of position-4 has been converted into ester prodrugs to increase bioavailability of cephalosporins, and these can be given orally as well. Examples include cefuroxime axetil and cefodoxime proxetil.
- Olefinic linkage at C 3-4 is essential for antibacterial activity. Isomerization of the double bond to 2-3 position leads to great losses in antibacterial activity.
Synthesis of 7-aminocephalosporonic acid from cephalosporin C
Cephalosporin C is isolated on an industrial scale by fermentation using Cephalosporium acremonium.
The key reaction, based on a method for removing glutamate residue in cephalosporin C, involves conversion of the primary amine in the molecule to a diazo function by reaction with nitrosyl chloride and formic acid. The diazo function can be displaced by oxygen from the enol form of amide at the 7-position to form iminolactone. Hydrolysis of the imine leads to 7-ACA.
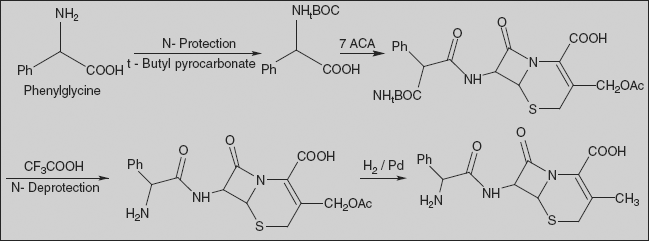
Cephalexin [6R-[6α,7β(R)]]-3-Methyl-8-oxo-7-[(aminophenylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Amino group of phenylglycine is protected with t-BOC, and activated carboxylic acid reacts with 7-ACA to give amide derivative. t-BOC is deprotected by treatment with trifluoro acetic acid. Reducing this product with hydrogen using a palladium on barium sulphate catalyst results in the deacetoxylation at the third position of 7-aminocephalosporanic acid, making the desired cephalexin.
Cefadroxil [6R-[6α,7β(R)]]-3-Methyl-8-oxo-7-[[amino(4-hydroxyphenyl)acetyl]amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
7-ACA is deacetylated by hydrogenolysis; carboxylate is protected with silyl derivative; and acylation with t-BOC-protected tyrosine and deprotection afford cefadroxil.
It is an analogue of cephalexin and differs only in the presence of a hydroxyl group in the fourth position of the phenyl ring of phenylglycine.
Cephradine [6R-[6α,7β(R)]]-3-Methyl-8-oxo-7-[(amino-1,4-cyclohexadien-1-ylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
It is a close analogue of cephalexin and differs in that the phenyl group in phenylglycine is partially hydrated to a 1,4-cyclohexadienyl moiety.
It is synthesised from phenylglycine, which is partially reduced by lithium in liquid ammonia, which forms 1,4-cyclohexadienylglycine, and the amino group in this compound is protected by reacting it with t-BOC. The resulting compound, on reaction with deacetoxylated 7-aminocephalosporanic acid, followed by deprotection gives cephradine.
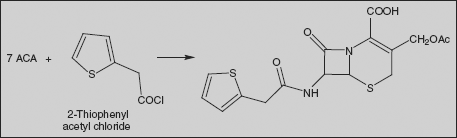
Cephalothin (6R-trans)-3-[(Acetyloxy)methyl]-8-oxo-7-[(2-thienylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Cephalothin is synthesised by direct interaction of 2-thienylacetic acid chloride with 7-aminocephalosporanic acid in the presence of sodium bicarbonate.
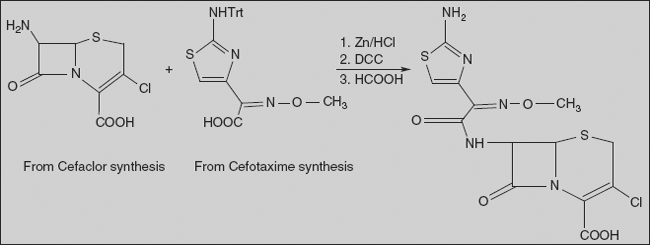
Cefaclor (6R,7R)-7-[(R)-2-amino-2-phenylacetamido]-3-chloro-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Penicillin V on treatment with peracetic acid gives sulphoxide, and carboxyl group is protected as 4-nitrobenzyl (PNB) ester. Reaction of this compound with a chlorinating agent such as N-chlorosuccinimide in the presence of acid results in chlorination on sulphur to form chlorosulphonium chloride; this compound then ring opens with the loss of hydrogen chloride to the unsaturated sulphonyl chloride. Treatment of this with Lewis acid leads to Friedel-Crafts-like attack on the olefin to afford a 6-membered cyclized product; the double bond concomitantly shifts to the exocyclic position. Ozonization followed by cleavage of the ozonide affords ring ketone; this exists as conjugated enol; sulphoxide is then reduced to cephem. Reaction of this with phosphorous pentachloride converts the enol to chloride, which on hydrolysis gives 7-amino derivative. This is converted to cefaclor by reaction with phenylglycine by the standard protocol mentioned above.
Cefamandole 7-D-Mandelamido-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Cefamandole is synthesised from 7-aminocephalosporanic acid. Reaction of this with 1-methyl-1,2,3,4-tetrazol-5-thiol results in displacement of the acetoxy group to form the intermediate. This is then acylated with acid chloride from the dichloroacetyl ester of D-mandellic acid. The protecting group on the side-chain is then removed to afford cefamandole.
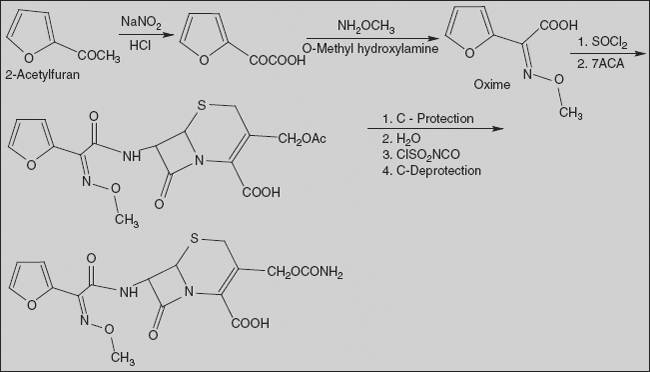
Cefuroxime (Z)-mono(O-methyloxim) (6R,7R)-7-[2-(2-furyl)glyoxylamido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid carbamate
It is synthesised from 2-acetylfuran. Oxidizing this compound with nitrous acid gives 2-furylglyoxalic acid, which is reacted with methoxylamine to give the corresponding oxime, syn-2-methoxyamino-2-(2-furyl)acetic acid. Acid is converted to acid chloride by reaction with thionyl chloride, which on reaction with 7-ACA gives amide. Carboxyl group is protected (with benzhydryl) and enzymatic hydrolysis in an alkaline medium, in which the benzhydryl protection is not affected, and only the acetoxy group of the molecule at position C3 of the aminocephalosporanic acid is hydrolysed. The resulting product with a free hydroxymethyl group is reacted with chlorosulphonyl isocyanate, with intermediate formation of the corresponding N-chlorosulphonyl urethane, which is hydrolysed by water to the urethane. Finally, removal of the benzhydryl protection using trifluoroacetic acid gives the desired cefuroxime.
Cefotaxime α-O-Methyloxime acetate (6R, 7R)-7-[2-(2-amino-4-thiazolyl)-glyoxylamido]-3-(hydroxy-methyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Cefotaxime is synthesised by acylating of 7-aminocephalosporanic acid with 2-(2-amino-4-thiazolyl)-2-methoxyiminoacetic acid, which is protected at the amino group by a trityl protection. After removing the trityl protection from the resulting product with dilute formic acid, the desired cefotaxime is formed. The ethyl ester of 2-(2-amino-4-thiazolyl)-2-methoxyiminoacetic acid necessary for this synthesis, as well as for the synthesis of a number of other antibiotics of the cephalosporin series, is synthesised from acetoacetic ester. Nitrosation of acetoacetic ester with nitrous acid gives isonitrosoacetoacetic ester. O-Methylation of the hydroxyl group of obtained product with dimethylsulphate in the presence of potassium carbonate gives ethyl 2-(methoxyimino) acetoacetate. Brominating the resulting product with bromine in methylene chloride in the presence of 4-toluenesulphonic acid gives 4-bromo-2-methoxyiminoacetoacetic ester. Reacting this with thiourea according to the classic scheme of preparing of thiazoles from α-bromocarbonyl compounds and thioamides gives the ethyl ester of 2-(2-amino-4-thiazolyl)-2-methoxyiminoacetic acid. Reacting this with triphenylchloromethane in the presence of triethylamine results in a trityl protection of the amino group, forming the ethyl ester of 2-(2-tritylamino-4-thiazolyl)-2-methoxyiminoacetic acid, which is hydrolysed to the acid using sodium hydroxide. The resulting acid, as was already stated, is used for acylating of 7-aminocephalosporanide acid in the presence of dicyclohexylcarbodiimide, giving tritylated cefotaxime, α-O-methyloxime acetate 7-[2-(2-tritylamino)-4-thiazolyl-glycoxylamido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid. Finally, removing the trityl protection from the synthesised product using dilute formic acid gives cefotaxime.
Ceftizoxime α-O-Methyloxime of (6R,7R)-7-[2-(2-amino-4-thiazolyl)glyoxylamido]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Acylation of 6-ACA derivative with thiazole derivative in presence of DCC, followed by deprotection of trityl group, affords ceftizoxime.
Ceftazidime 1-[[7-[[(2-Amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]pyridin-2-carboxylic acid
Reaction between ethyl 2-[2-(tritylamino)-1,3-thiazol-4-yl]-2-hydroxyiminoacetate and ethyl 2-bromo-2-methylpropanoate gives O-alkylated compound, and ethoxycarbonyl group in this molecule is hydrolysed by sodium hydroxide to acid. The acid is activated with DCC and amide formed by reaction with 7-ACA. Formic acid treatment removes the trityl group from amino group, and is followed by treatment with pyridine to replace the acetoxyl group with a pyridine group to afford ceftazidime.
Cefoperazone (6R,7R)-7-[(R)-2-(4-Ethyl-2,3-dioxo-1-piperazincarboxamido)-2-(p-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid
Reaction between 4-ethylpiperazin-2,3-dion-1-carboxylic acid chloride and the sodium salt of 4-hydroxyphenylglycine gives acid derivative. The acid is activated with DCC and amide formed by reaction with 7-ACA; this on reaction with 1-methyl-1,2,3,4-tetrazol-5-thiol results in displacement of the acetoxy group to form cefoperazone.
26.2 TETRACYCLINE ANTIBIOTICS
Tetracyclines are potent, broad-spectrum antibacterial agents effective against a host of gram-positive and gram-negative aerobic and anaerobic bacteria. As a result, the tetracyclines are drugs of choice or well-accepted alternatives for a variety of infectious diseases. Among these, their role in the treatment of sexually transmitted and gonococcal diseases, urinary tract infections, bronchitis, and sinusitis remains prominent.
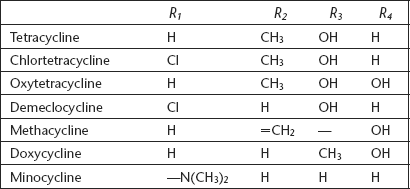
The majority of the marketed tetracyclines (tetracycline, chlortetracycline, oxytetracycline, and demeclocycline) are naturally occurring compounds obtained from fermentation of Streptomyces spp broths. The semi-synthetic tetracyclines (methacycline, doxycycline, minocycline) have an advantage of longer duration of antibacterial action. However, all these tetracyclines exhibit a similar profile in terms of antibacterial potency. In general, the activity encompasses many strains of gram-negativeE.coli, Proteus, Klebsiella, Enterobacter, Niesseria, and Serratia spp. as well as gram-negativeStreptococci and Staphylococci. Of particular interest is the potency of tetracyclines againstHaemophilus, Legionella, Chlamydia, and Mycoplasma.
Mechanism of action: The tetracyclines exhibit bacteriostatic effects on growing bacteria via the inhibition of protein synthesis. Their action occurs at the ribosomal level, where the drug binding to the 30S ribosomal subunit takes place on the ribosome-mRNA complex. This phenomenon stops the attachment of amino acylated t-RNA molecules and prevents peptide-chain growth.
26.2.1 Structural Features and SAR of Tetracyclines
The key structural feature is a linearly fused tetracyclic nucleus, and each ring needs to be six-membered and purely carbocyclic. The D-ring needs to be aromatic and the A-ring must be appropriately substituted at each of its carbon atoms for notable activity. The B-ring and the C-ring tolerate certain substituent changes as long as the keto-enol system (at C-11, 12, 12a) remains intact and conjugated to the phenolic D-ring. The D-, C-, B- ring phenol-, keto-, enol system is imperative, and the A-ring must also contain a conjugated keto-enol system. Specifically, the A-ring contains a tricarbonyl-derived keto-enol array at positions C-1, 2, and 3. Other structural requirements for good antibacterial activity include a basic amine function at the C-4 position of the A-ring.
C-1 substituents: The keto-enol system of the A-ring is indispensable for antibacterial activity. No variation at the C-1 position has been successful.
C-2 substituents: The carboxamide moiety is present in all naturally occurring tetracyclines and this group is crucial for antibacterial activity. The amide is best left unsubstituted, or mono-substitution is acceptable in the form of activated alkylaminomethyl amide (Mannich bases). An example is rolitetracycline. Large alkyl group on the carboxamide may alter the normal keto-enol equilibrium of the C-1, 2, and 3 conjugated system, and diminishes inherent antibacterial activity. The replacement of carboxamide group or dehydration of carboxamide to the corresponding nitrile results in the loss of activity.
C-3 substituents: In conjugation with the C-1 position, the keto-enol conjugated system is imperative for antibacterial activity.
C-4 substituents: The naturally occurring tetracyclines contain α-C-4 dimethylamino substituent that favourably contributes to the keto-enolic character of the A-ring. Replacement of dimethylamino group with a hydrazone, oxime, or hydroxyl group leads to a pronounced loss of activity, probably due to the increase in heteroatom basicity.
C-4a substituents: The α-hydrogen at C-4a position of tetracyclines is necessary for useful antibacterial activity.
C-5 substituents: Many naturally occurring antibacterial tetracyclines have an unsubstituted methylene moiety at the C-5 position. However, oxytetracycline contains C-5 α-hydroxyl group and was found to be a potent compound, and has been modified chemically to some semi-synthetic tetracyclines. Alkylation of the C-5 hydroxyl group results in a loss of activity.
Ester formation is only acceptable if the free oxytetracycline can be liberated in vivo; only small alkyl esters are useful.
C-5a substituents: The configuration of the naturally occurring tetracyclines places the C-5a hydrogen atom in an α-configuration. Epimerization is detrimental to antibacterial activity.
C-6 substituents: The C-6 position is tolerant of a variety of substituents. The majority of tetracyclines have an α-methyl group and a β-hydroxyl group at this position. Demeclocyclin is a naturally occurring C-6 demethylated chlortetracycline with an excellent activity. This C-6 methyl group contributes little to the activity of tetracycline. Similarly, the C-6 hydroxyl group also appears to offer little in terms of antibacterial activity; removal of this group affords doxycycline, which is a superb antibacterial.
C-7 and C-9 substituents: The nature of the aromatic D-ring predisposes the C-7 position to electrophilic substitution, and nitro and halogen groups have been introduced. Some C-7 nitro tetracyclines are among the most potent of all tetracyclines in vitro, but these compounds were potentially toxic/carcinogenic. Halogenated derivatives are less active. The C-7 acetoxy, azido, and hydroxyl tetracyclines are inferior in terms of antibacterial activity.
C-10 substituents: The C-10 phenolic moiety is absolutely necessary for antibacterial activity.
C-11 substituents: The C-11 carbonyl moiety is part of one of the conjugated keto-enol systems required for antibacterial activity.
C-11a substituents: In general, few modifications at the C-11a position of tetracycline have been tolerated. This is probably due to the detrimental effects exerted upon the keto-enol system, which is vital for magnesium cation binding and subsequent tetracycline uptake by the bacterial cell.
C-12 substituents: As with the C-11 position, the C-12 position is part of the keto-enol system vital for drug uptake, binding, and observed antibacterial activity.
C-12a substituents: The C-12a hydroxyl group is needed for antibacterial activity, although this moiety can be esterified to provide tetracycline with increased lipophilicity. Antibacterial properties are retained if the alkyl ester is small in size, and readily undergoes hydrolysis to liberate free tetracycline.
26.2.2 Structure of Tetracyclines
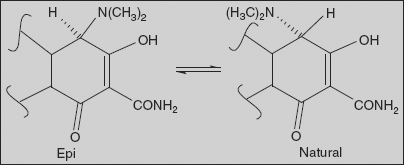
Effect of pH on tetracyclines An interesting property of tetracyclines is their ability to undergo epimerization at C-4 in solutions of intermediate pH range. These isomers are called epitetracyclines. Under the influence of the acidic conditions, an equilibrium is established in about one day consisting of approximately equal amount of isomers. Epitetracyclines exhibit much less activity than natural isomers.
Strong acids and bases attack the tetracyclines having a hydroxyl group on C-6, causing a loss in activity through modification of C-ring. Strong acids produce dehydration through a reaction involving the C-6 hydroxyl group and C-5a hydrogen. The double bond formed between positions C-5a and C-6 induces a shift in the position of double bond between C-11a and C-12 to a position between C-11 and C-11a, forming the more energetically favoured resonance of the naphthalene group found in the inactive anhydrotetracyclines.
Bases promote a reaction between the C-6 hydroxyl group and the ketone group at the C-11 position, causing the bond between the C-11 and C-11a atoms to cleave and to form the lactone ring found in the inactive isotetracycline.
Tetracyclines can stain developing teeth (even when taken by the mother during pregnancy). Inactivated by Ca2+ ion, these are not to be taken with milk or yogurt. Inactivated by aluminium, iron, and zinc, these are not to be taken at the same time as indigestion remedies. These are inactivated by common antacids and over-the-counter heartburn medicines. These should be avoided during pregnancy as it may affect bone growth of foetus.
Effect of metals on tetracycline Stable chelate complexes are formed by tetracycline with many metals, including calcium, magnesium, and iron. Such chelates are insoluble in water, accounting for impairment in absorption of most tetracyclines in the presence of milk, calcium, magnesium, and aluminium-containing antacids and iron salts.
The affinity of tetracycline for calcium causes them to be laid down in newly formed bones and teeth as tetracycline-calcium orthophosphate complexes. Deposits of these antibiotics in teeth cause a yellow discolouration that darkens because of photochemical reaction. Tetracyclines are distributed into the milk of lactating mothers and also cross the placenta into the foetus. The possible effect of these agents on bones and teeth of the child should be taken into consideration before their use in pregnancy or in children under eight years of age.
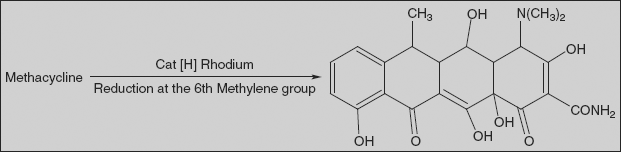
Methacycline 4-Dimethylamino-1,4,4a,5,5a,6,11,12a-octahydro-3,6,10,12,12a-pentahydroxy-6-methylen-1,11-dioxo-2-naphthacencarboxamide
Synthesis
It is synthesised from oxytetracycline, which is reacted with a sulphur trioxide-pyridine complex, resulting in an oxidation reaction. Simultaneous sulphonation gives a naphthacen-sulphotetrahydrofuran derivative intermediate, which when reacted with hydrofluoric acid forms methacycline.
Doxycycline 4-Dimethylamino-1,4,4a,5,5a,6,11,12a-oxtahydro-3,5,10,12,12a-pentahydroxy-6-methyl-1,11-dioxo-2,naphthacencarboxamide
Doxycycline is an isomer of tetracycline that differs only in the placement of one hydroxyl group. Doxycycline can be formally viewed as the result of transferring the C6 hydroxyl group of tetracycline to C5.
Synthesis
Doxycycline is synthesised in two different ways; one of the ways suggests dehydrating oxytetracycline at C6 by reducing the tertiary hydroxyl group with hydrogen using a rhodium on carbon catalyst. It is also synthesised by reducing methylene group of methacycline using rhodium on carbon catalyst.
Minocycline 4,7-bis(Dimethylamino)-1,4,4a,5,5a,6,11,12 α-octahydro-3,10,12,12a-tetrahydroxy-1,11-dioxo-2-naphthacencarboxamide
Synthesis
Hydrogenolysis of demethylchlortetracycline removes both the chlorine and the benzylic hydroxyl group. The product, 6-demethyl-6-deoxytetracycline, on nitration gives mixture of 7 (a)- and 9 (b)-nitro-6-demethyl-6-deoxytetracycline; (a) is reductively methylated by catalytic hydrogenation in the presence of formaldehyde to give minocycline.
Rolitetracycline (2Z,4S,4aS,5aS,6S,12aS)-4-Dimethylamino-6,10,11,12a-tetrahydroxy-2-[hydroxy-(pyrrolidin-1-ylmethylamino)methylidene]-6-methyl-4,4a,5,5a-tetrahydrotetracene-1,3,12-trione
It is a water-soluble prodrug of tetracycline.
Synthesis
It is prepared by reacting tetracycline, formaldehyde, and pyrrolidine; amino methylation takes place in amide group (Mannich reaction).
26.3 MACROLIDE ANTIBIOTICS
The macrolide antibacterial agents are extremely useful chemotherapeutic agents for the treatment of a variety of infectious disorders and diseases caused by a host of gram-positive bacterial pathogens. These agents as exemplified by erythromycins are generally effective against Streptococci, Staphylococci, Chlamydia, Legionella, and Mycoplasma. As a result, the macrolides are commonly administered for respiratory, skin and tissue, and genitourinary infections caused by these pathogens.
Chemistry The macrolide antibiotics have three common chemical characteristics:
- A large lactone ring
- A ketone group
- A glycosidically linked amino sugar
Usually, the lactone ring has 12, 14, or 16 atoms in it and is often partially unsaturated, with an olefinic group conjugated with a ketone function. They may have, in addition to the amino sugar, a neutral sugar that is glycosidically linked to the lactone ring.
R
|
R1
| |
Erythromycin
|
O
|
H
|
Roxithromycin
|
CH3OCH2CH2OCH2ON ======
|
H
|
Clarithromycin
|
O
|
CH3
|
Mecahnism of action The macrolides exert their antibacterial effects, which are usually bacteriostatic, via inhibition of bacterial protein biosynthesis. Specifically, the macrolides target the 50S ribosomal subunit; different members stop protein synthesis at varying stages of peptide-chain elongation. The macrolides inhibit ribosomal peptidyl transferase activity. Some macrolides also inhibit the translocation of the ribosome along the m-RNA template.
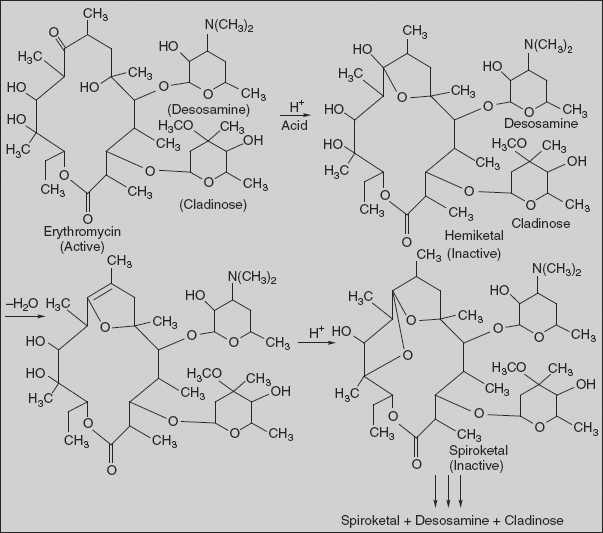
Acid degradation of erythromycin [(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-Cmethyl-3-O-methyl-α-L-ribo-hexopyranosyl)-oxy]-14-ethyl-7,12,13-trihydroxy-3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecan-2,10-dione]:
Erythromycin is unstable in acid media. The C-6 hydroxyl group reversibly attacks the C-9 ketone, giving rise to a hemiketal intermediate. Dehydration prevents regeneration of the parent erythromycin, and the C-12 hydroxyl group can subsequently add to produce a spiroketal species. The cladinose group is cleaved from the macrocycle and more harsh conditions lead to the release of desosamine. Useful antibacterial activity lasts upon dehydration of the hemiketal, and the spiroketal is also weakly active.
26.3.1 SAR of Macrolide Antibiotics
- A number of strategies have been utilized to improve the acid stability of erythromycin.
- The first approach involved the addition of hydroxylamine to the ketone to form oxime— e.g., roxithromycin.
- The second approach involves an alteration of C-6 hydroxyl group, which is the nucleophilic functionality that initiates erythromycin degradation. Modification that removes the nucleophilic nature of this hydroxyl group can retain antibacterial properties if the size of the group is kept small so as not to affect the ribosomal binding—e.g., clarithromycin.
- The azalides (e.g., azithromycin) are semi-synthetic 15-membered congeners in which a nitrogen atom has been introduced to expand a 14-membered precursor, and this leads to an extended spectrum of action.
Synthesis
Roxithromycin (3R,4S,5S,6R,7R,9R,11S,12R,13S,14R)-6-[(2S,3R,4S,6R)-4-dimethylamino-3-hydroxy-6-methyloxan-2-yl]oxy-14-ethyl-7,12,13-trihydroxy-4-[(2R,4R,5S,6S)-5-hydroxy-4-methoxy- 4,6-dimethyloxan-2-yl]oxy-10-(2-methoxyethoxymethoxyimino)-3,5,7,9,11,13-hexamethyl-1-oxacyclo-tetradecan-2-one
Roxithromycin is derived from erythromycin, containing the same 14-membered lactone ring. However, an N-oxime side-chain is attached to the lactone ring.
It is prepared by reacting erythromycin with CH3OCH2CH2OCH2ONH2 (substituted hydroxylamine).
Clarithromycin (2R,3S,4S,5R,6R,8R,10R,11R,12S,13R)-3-(2,6-dideoxy-3-C-3-O-dimethyl-α-L-ribo-hexopyranosyloxy)-6-methoxy-9-oxo-11,12-dihydroxy-2,4,6,8,10,12-hexamethyl-5-(3,4,6-trideoxy-3-dimethylamino-β-D-xylo-hexopyranosyloxy)cyclopentadecan-13-olide.
It is a semi-synthetic analogue of erythromycin A, in which the hydroxyl group at C6 is replaced with a methoxyl group. It is prepared by selective methylation of 6-OH group of erythromycin.
Azithromycin [2R-(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-3-(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyloxy)-2-ethyl-3,4,10-trihydroxy-3,5,6,8,10,12,14-heptamethyl-11-[(3,4,6-trideoxy-3-dimethylamino)-β-D-xylo-hexopyranosyl]-oxy]-1-oxa-6-azacyclopentadecan-15-one.
It differs chemically from erythromycin in that a methyl-substituted nitrogen atom is incorporated into the lactone ring, thus making the lactone ring 15-membered.
Synthesis
It is prepared from erythromycin by reacting it with hydroxylamine to give oxime derivative, which undergoes Beckmann rearrangement to form ring-enlarged amide derivative. Wolff-Kishner reduction of amide to amine followed by N-methylation with methyl iodide affords azithromycin.
Unlike erythromycin, azithromycin is acid-stable and can, therefore, be taken orally with no need of protection from gastric acids. It is readily absorbed, and its absorption is greater on an empty stomach.
26.4 AMINOGLYCOSIDE ANTIBIOTICS
The aminoglycosides each contains one or more amino sugars, such as glucosamine or neosamine, linked by glycosidic linkages to a basic (amino or guanidine) six-membered carbon ring. These are broad-spectrum antibiotics, and in general they have greater activity against gram-negative than gram-positive bacteria.
Toxicity The toxicity of these agents is dose-related, and therefore every individual can get these side-effects provided the dose is sufficiently high enough. Because of their potential for ototoxicity and nephrotoxicity (kidney toxicity), aminoglycosides are administered in doses based on body weight. Vestibular damage, hearing loss, and tinnitus are irreversible, so care must be taken not to achieve a sufficiently high dose. Concomitant administration of a cephalosporin may lead to increased risk of nephrotoxicity, while administration with a loop diuretic increases the risk of ototoxicity. These properties have limited the use of aminoglycoside chemotherapy to serious systemic indications. Some aminoglycosides can be administered for ophthalmic and topical purposes.
Mechanism of action: Aminoglycosides work by binding to the bacterial 30S ribosomal subunit, inhibiting the translocation of the peptidyl-tRNA from the A-site to the P-site and also causing misreading of mRNA, leaving the bacterium unable to synthesise proteins vital to its growth.
Examples
Name
|
Source
|
1. Gentamycin
|
Micromonospora purpurea
|
2. Neomycin
|
Streptomyces fradiae
|
3. Streptomycin
|
Streptomyces griseus
|
4. Tobramycin
|
Streptomyces tenebrarius
|
5. Framycetin
|
Streptomyces decaris
|
6. Kanamycin
|
Streptomyces kanamyceticus
|
7. Amikacin
|
It is 1-L-(-) 4-amino-2-hydroxybutyryl kanamycin
|
8. Capreomycin
|
Streptomyces capreolus
|
26.5 MISCELLANEOUS ANTIBIOTICS
Chloramphenicol D-threo-2,2-Dichloro-N-[β-hydroxy-α-(hydroxymethyl)]-N-nitrophenylacetamide
It is also obtained from Streptomyces venezulae. Because of its side-effect (blood dyscrasias) and availability of safer agents, the use of this agent declined.
Chloramphenicol inhibits protein synthesis in bacteria, and to a lesser degree in eukaryotic cells. It easily diffuses into the bacterial cell, where it reversibly binds with the 50S ribosomal subunit. This prevents the amino acid ending of tRNA from binding with the binding regions of the 50S ribosome. The binding of aminoacyl tRNA with the 30S subunit is not disturbed. Mammalian cells containing 80S ribosomes are not affected by chloramphenicol. However, this drug inhibits synthesis of mitochondiral proteins in mammalian cells, possibly because of the similarity between mitochondrial and bacterial ribosomes.
Synthesis
The starting material is prepared by aldol condensation of benzaldehyde with 2- nitroethanol to give four enantiomers of nitropropanediol; the total mixture is reduced to the corresponding mixture of aminodiols. The threo isomer is then separated by crystallization and resolved as a diasteromeric salt to give D(–) isomer. Acylation with dichloroacetyl chloride gives triacetate, which on saponification gives the desired amide derivative. The free hydroxyl groups are then acylated with acetic anhydride and the product is nitrated at 4th position. Saponification of ester gives chloramphenicol.
Bacitracin It is a polypeptide antibiotic obtained from Bacillus subtilis. Bacitracin interferes with the dephosphorylation of the C55-isoprenyl pyrophosphate, a molecule that carries the building blocks of the peptidoglycan bacterial cell wall outside of the inner membrane. Bacitracin does not work well orally. However, it is very effective topically. Ten individual bacitracins have been isolated: bacitracins A, A1, B, C, D, E, F1, F2, F3, and G. However, the drug itself, named bacitracin, that is used in medicine is a mixture of polypeptide antibiotics.
Polymyxin B sulphate Polymyxines are a group of related polypeptide antibiotics that are produced by spore-forming soil bacteria Bacillus polymyxa and B. circulans, and they differ in amino acid content. Five different polymyxines have been identified - polymyxines A, B, C, D, and E, which differ in the amino acid content.
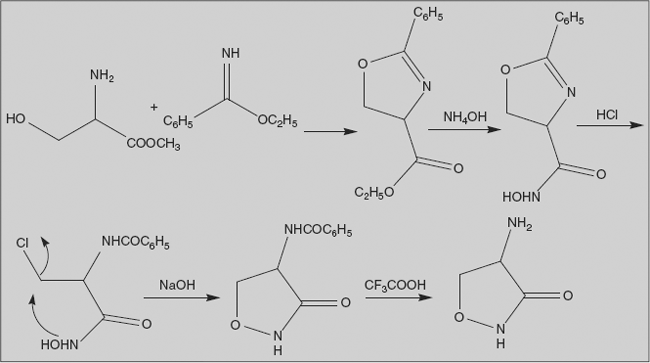
Cycloserine 4-Aminoisoxazolidin-3-one
It is obtained from Streptomyces orchidaceus. Cycloserine is an antibiotic effective againstMycobacterium tuberculosis. The terminal two amino acid residues of the murein precursor lipid II consist of D-alanine, which is produced by the enzyme alanine racemase; the two residues are joined by D-alanine ligase. Both enzymes are competitively inhibited by cycloserine.
Synthesis
Protection of serine methyl ester as its isoxazoline is attained by reaction with the iminoether obtained from benzonitrile. Ester-amide interchange with hydroxylamine in presence of base gives hydroxamic acid. Reaction of this with hydrogen chloride leads to ring opening with concurrent conversion of hydroxyl group to chloride. Base converts this hydroxamic acid to its anion; the negatively charged oxygen then displaces chlorine to form isoxazoline. Removal of benzoyl group affords cycloserine.
26.6 NEWER DRUGS
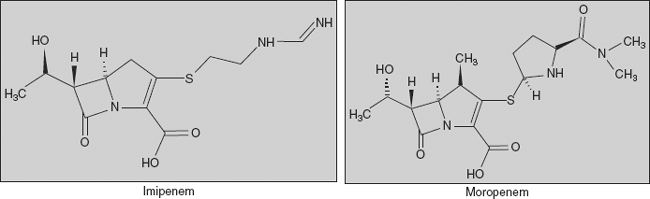
Carbapenems Carbapenems are a class of β-lactam antibiotics with a broad spectrum of antibacterial activity, and have a structure that renders them highly resistant to β-lactamases. The carbapenems are structurally very similar to the penicillins, but the sulphur atom in position 1 of the structure has been replaced with a carbon atom, and hence the name of the group, the carbapenems. Examples:
(a) Imipenem- ((5R,6S)-3-[2-(aminomethylideneamino)ethylsulphanyl]-6-(1-hydroxyethyl)-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid) and (b) moropenem (3-[5-(dimethylcarbamoyl) pyrrolidin-2-yl] sulphanyl-6- (1-hydroxyethyl)-4-methyl-7-oxo- 1-azabicyclo[3.2.0] hept-2-ene-2-carboxylic acid)
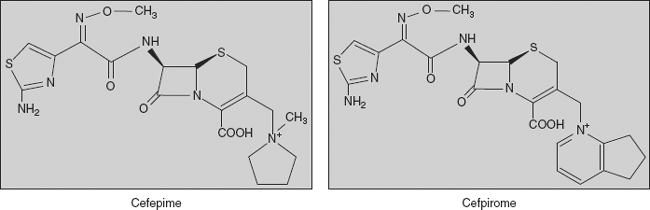
Fourth-generation cephalosporins Broad spectrum with enhanced activity against gram-positive bacteria and beta-lactamase stability
Monobactams: Unlike other beta-lactams, the monobactam contains a nucleus with no fused ring attached. Thus, there is less probability of cross-sensitivity reactions.
Example:
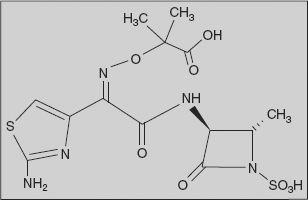
Aztreonam: (3-[2-(2-azaniumyl-1,3-thiazol-4-yl)-2-(1-hydroxy-2-methyl-1-oxo-propan-2-yl)oxyimino-acetyl]amino-2-methyl-4-oxo-azetidine-1-sulphonate)
β-Lactamase inhibitors: Although they exhibit negligible antimicrobial activity, they contain the β-lactam ring. Their sole purpose is to prevent the inactivation of β-lactam antibiotics by binding the β-lactamases, and, as such, they are co-administered with β-lactam antibiotics. These drugs are irreversible inhibitors of β-lactamase; these bind the enzyme and do not allow it to interact with the antibiotic.
Example:
Teicoplanin: It is an antibiotic used in the prophylaxis and treatment of serious infections caused by gram-positive bacteria. It is a glycopeptide antiobiotic extracted from Actinoplanes teichomyceticus. Its mechanism of action is to inhibit bacterial cell-wall synthesis. Teicoplanin is actually a mixture of several compounds, five major (named teicoplanin A2-1 through A2-5) and four minor (named teicoplanin RS-1 through RS-4). All teicoplanins share a same glycopeptide core, termed teicoplanin A3-1 - a fused ring structure to which two carbohydrates (mannose and N-acetylglucosamine) are attached. The major and minor components also contain a third carbohydrate moiety β-D-glucosamine, and differ only by the length and conformation of a side-chain attached to it.
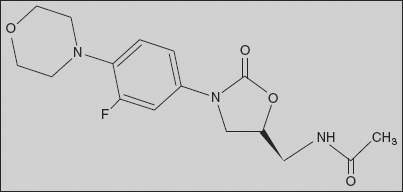
Linezolid: N-[[3-(3-Fluoro-4-morpholinophenyl)-aoxooxazolidin-5-yl]methyl]acetamide
It is an antibiotic drug. It was the first commercially available oxazolidinone antibiotic and is usually reserved for the treatment of serious bacterial infections where older antibiotics have failed due to antibiotic resistance. The drug works by inhibiting the initiation of bacterial protein synthesis; it is the only antibiotic to work in this manner. Linezolid is effective against gram-positive pathogens, notably Enterococcus faecium, Staphylococcus aureus, Streptococcus agalactiae, Streptococcus pneumoniae, and Streptococcus pyogenes. It has almost no effect on gram-negative bacteria and is only bacteriostatic against most Enterococcus species.