Free pharmacy material























Local Anaesthetics
INTRODUCTION
Local anaesthetics are medications used for the purpose of temporary and reversible elimination of painful feelings in specific areas of the body by blocking transmission of nerve fibre impulses. These drugs, unlike general anaesthetics, cause a loss of feeling in specific areas while keeping the patient conscious.
Local anaesthetics are used for pain relief, soreness, itching, and irritation associated with disturbance of the integrity of the skin and mucous membranes (cuts, bites, wounds, rashes, allergic conditions, fungal infections, skin sores, and cracking). They are used during opthalmological procedures such as tonometry and gonioscopy, removal of foreign bodies, and minor surgical interventions. Local anaesthetics are widely used in surgery, gynaecology, and dentistry. In certain cases, local anaesthetics (lidocaine, procainamide) can be used as anti-arrhythmic drugs.
IDEAL PROPERTIES OF LOCAL ANAESTHETICS
- Non-irritating to tissues and not causing any permanent damage
- Low systemic toxicity
- Effective whether injected into the tissue or applied locally to skin or mucous membranes
- Rapid onset of anaesthesia and short duration of action
MECHANISM OF ACTION
A mechanism of local anaesthetic action in which they serve as sodium channel blockers has been proposed. According to this mechanism, the molecular targets of local anaesthetic action are the voltage-requiring sodium channels, which are present in all the neurons. The process of local anaesthesia by respective drugs can be represented in the following manner.
In a resting condition, there is a specific rest potential between the axoplasm and the inner parts of the cell. This rest potential is maintained by relative concentration of sodium and potassium ions along the membrane of the nerve. During nerve stimulation, the membrane is depolarised and sodium channels in that area are opened, allowing sodium ions to rush into the cell. At the peak of depolarization, potassium channels are opened. The last ones leave the cell and the cell is repolarised.
This process lasts 1–2 msec, after which the nerve cell, having transmitted the necessary impulse, restores its ion gradient.
It is believed that after introduction of local anaesthetic into the organism in the form of a water-soluble salt, equilibrium is established between the neutral and cationic forms of the used drug depending on the pKa of the drug and the pH of the interstitial fluid. It is also believed that only the uncharged (neutral) drug form can pass through – it passes through connective tissue surrounding the nerve fibre and through the phospholipid plasma membrane into the axoplasm. In the axoplasm, the base is once again ionised until it reaches an appropriate value determined by intracellular pH.
It is suspected that these drugs selectively bind with the intracellular surface of sodium channels and block the entrance of sodium ions into the cell. This leads to stoppage of the depolarization process, which is necessary for the diffusion of action potentials, elevation of the threshold of electric nerve stimulation, and thus the elimination of pain. Since the binding process of anaesthetics to ion channels is reversible, the drug diffuses into the vascular system where it is metabolised, and nerve cell function is completely restored.
6.4 CLINICAL USES OF LOCAL ANAESTHETICS
Local anaesthesia is the loss of sensation in the body part without the loss of consciousness or the impairment of central control of vital functions, used for minor surgical procedures.
Local anaesthetics are categorised by the method of administration.
- Topical anaesthesia: Anaesthesia of mucous membranes of the nose, throat, tracheobronchial tree, oesophagus, and genito-urinary tract can be produced by direct application of aqueous solutions of salts of many local anaesthetics or by suspension of the poorly soluble local anaesthetics. For prolonged duration of action, vasoconstriction can be achieved by the addition of a low concentration of a vasoconstrictor such as phenylephrine (0.005%): e.g., lignocaine (2–10%), cocaine (1–4%), and tetracaine (2%).
- Infiltration anaesthesia: In this, local anaesthetics are injected directly into the tissue, which may be superficial tissue of the skin or deeper structures including intra-abdominal organs. The duration of infiltration anaesthesia can be doubled by the addition of epinephrine (5 µg/ml) to the injection solution: e.g., lignocaine (0.5–1.0%), procaine (0.5–1.0%), and bupivacaine (0.125–0.25%).
- Field block anaesthesia: It is produced by subcutaneous injection of a local anaesthetics solution in such a manner as to anaesthetise the region distal to the injection. The advantage of field block anaesthesia is that less drug can be used to provide a greater area of anaesthesia than when infiltration anaesthesia is used: e.g., lignocaine (0.5–1.0%), procaine (0.5–1.0%), and bupivacaine (0.125–0.25%).
- Nerve block anaesthesia: This involves injection of a solution of local anaesthetics into or about individual peripheral nerves or nerve plexus: e.g., lignocaine (1.0–1.5%), mepivacaine (up to 7mg/kg of 1.0–2.0%), and bupivacaine (2–3 mg/kg of 0.25–0.375%). Addition of 5 µg/ml epinephrine prolongs duration.
- Intravenous regional anaesthesia: This technique relies on using the vasculature to bring the local anaesthetics solution to the nerve trunks and endings. It is used most often for the forearm and hand, but can also be adapted for the foot and distal leg: e.g., lignocaine (0.5%) and procaine (0.5%).
- Spinal anaesthesia: It follows the injection of local anaesthetics into the cerebrospinal fluid in the lumbar space: e.g., lignocaine, tetracaine, and bupivacaine.
- Epidural anaesthesia: In this, local anaesthetics is injected into epidural space – the space bounded by the ligamentum flavum posteriorly, the spina periosteum laterally, and the dura anteriorly: e.g., bupivacaine (0.5–0.75%), etidocaine (1.0–1.5%), lignocaine (2%), and chloroprocaine (2–3%).
CLASSIFICATION OF LOCAL ANAESTHETICS
1. Benzoic acid derivatives
2. p-Aminobenzoic acid derivatives
3. Anilides
4. Miscellaneous: Phenacaine, Diperodon, Dimethisoquin, Pramoxine, Dyclonine, Dibucaine
5. Newer drugs: Ropivacaine, Levobupivacaine
STRUCTURES, SYNTHESIS, AND STRUCTURE-ACTIVITY RELATIONSHIP (SAR) OF BENZOIC ACID DERIVATIVES
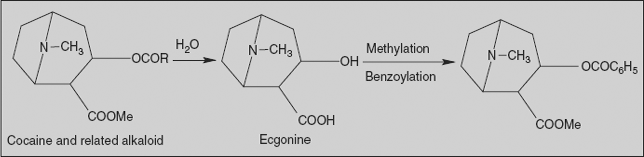
Cocaine
(-)3-(Benzoyloxy)-8-methyl-8-azabicyclooctane-2-carboxylic acid methyl ester
Synthesis
Commercial production involves total extraction of cocaine and related alkaloidal bases from the leaves of Erythroxylon coca, followed by acid hydrolysis of the ester alkaloid to obtain the total content of (−) ecgonine. After purification of ecgonine, cocaine is synthesised by esterification with methanol and benzoic acid.
Hexylcaine Hydrochloride
1-(Cyclohexylamino)-2-propanol benzoate
Synthesis
Reductive alkylation of amino alcohol with cyclohexanone affords the secondary amine. Acylation with benzoyl chloride affords hexylcaine.
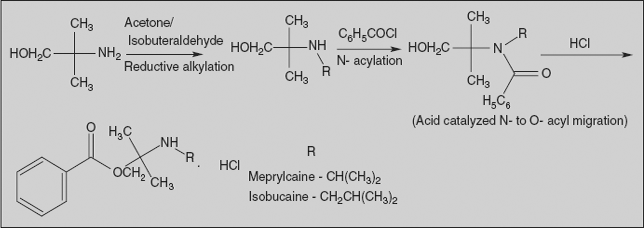
6.6.3 Meprylcaine and Isobucaine Hydrochloride
2-Methyl-2-(propylamino/isobutylamino)-1-propanol benzoate
Synthesis
Reductive alkylation of amino alcohol with acetone/isobutyraldehyde affords the corresponding amines. Acylation of the amine with benzoyl chloride probably goes initially to the amide. The acid catalysis used in the reaction leads to an N- to O-acyl migration to afford corresponding compounds.
Cyclomethycaine Sulphate
3-(2-Methylpiperidino)propyl-p-(cyclohexyloxy) benzoate sulphate
Synthesis
O-alkylation of p-hydroxy benzoic acid with cyclohexyl iodide affords the cyclohexyl ether under alkaline reaction condition. The acid group is activated with thionyl chloride and the acylation of the acid chloride with alcohol affords cyclomethycaine.
Piperocaine Hydrochloride
3-(2-Methylpiperidino)propyl benzoate
Synthesis
Benzoylation of 3-chloropropan-1-ol affords the haloester. Condensation of this intermediate with the reduction product of α-picoline affords piperocaine.
STRUCTURES, SYNTHESIS, AND SAR OF P-AMINOBENZOIC ACID DERIVATIVES
Benzocaine
Ethyl-p-aminobenzoate
Synthesis
Toluene on nitration with nitrating mixture affords 4-nitro toluene, which on oxidation followed by esterification yields ethyl ester derivative. Nitro group is reduced with tin and HCl affords benzocaine.
The mechanism of benzocaine action differs slightly from that mentioned above. It presumably acts by diffusing across the phospholipid membrane and then stretching it out. This deforms the sodium channels, which in turn—and in a unique manner—lowers sodium conduction.
Butamben
Butyl-p-aminobenzoate
Synthesis
4-Nitro benzoic acid on esterification with n-butanol followed by reduction affords butamben.
Procaine Hydrochloride
2-(Diethylamino)ethyl-p-aminobenzoate
Synthesis
Ethylene on reaction with hypochlorous acid yields ethylene chlorohydrin, which on reaction with diethylamine affords 2-(diethylamino) ethanol. Benzoylation of alcohol followed by reduction affords procaine.
Chloroprocaine
Chloroprocaine differs structurally from procaine in having a chlorine substituent in the 2-position of the aromatic ring. The electron-withdrawing chlorine atom destabilizes the ester group to hydrolysis. Chloroprocaine is hydrolysed by plasma more than four times faster than procaine. It is more rapid in onset of action and more potent than procaine. Chloroprocaine is used in situations requiring fast-acting pain relief.
Tetracaine
2-(Dimethylamino)ethyl-p-(butylamino)benzoate
Synthesis
Benzocaine undergoes alkylation with butyl bromide in presence of base; this is followed by trans-esterification reaction with 2-(diethylamino) ethanol, which affords tetracaine.
Butacaine
3-(Di-n-butylamino)-1-propanol-p-amino benzoate
Synthesis
It is similar to that of procaine, but uses n-dibutylamine instead of diethylamine.
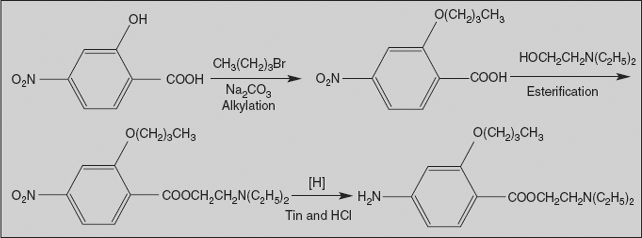
Benoxinate
2-(Diethylamino)ethyl-4-amino-2-butoxy benzoate
Synthesis
Benoxinate is prepared from 4-nitro salicylic acid by a three-step synthesis: first, by o-alkylation with n-butyl bromide; secondly, esterification with 2-(diethylamino) ethanol; and finally, reduction of nitro group affords benoxinate.
Propoxycaine
2-(Diethylamino)ethyl-4-amino-2-propoxy benzoate hydrochloride
Synthesis
It is similar to that of benoxinate, but alkylation is with n-propylbromide.
Proparacaine Hydrochloride
2-(Diethylamino)ethyl-3-amino-4-propoxy benzoate hydrochloride
Synthesis
It is similar to that of benoxinate, but starting material is 3-nitro salicylic acid and alkylation with propylbromide.
Structure-activity relationships (SAR) of benzoic acid derivatives
The benzoic acid derivatives are represented as follows:
Aryl group
- The clinically useful local anaesthetics of this series possess an aryl radical attached directly to the carbonyl group.
- Substitution of aryl group with substituents that increase the electron density of the carbonyl oxygen enhances activity.
- Favourable substituents in aryl ring include (electron-donating groups) alkoxy (propoxycaine), amino (procaine), and alkylamino (tetracaine) groups in the para or ortho positions. This homologous series increases partition coefficients with increasing number of methylene group (-CH2-). Local anaesthetics activity peaked with the C4-, C5-, or C6-homologous: e.g., tetracaine, cyclomethycaine.
- Aryl aliphatic radicals that contain a methylene group between the aryl radical and the carbonyl group result in compounds that have not found clinical use.
Bridge X
- The bridge X may be carbon, oxygen, nitrogen, or sulphur.
- In an isosteric procaine series, anaesthetic potency decreased in the following order: sulphur, oxygen, carbon, nitrogen.
- These modifications also affect duration of action and toxicity. In general, amides (X=N) are more resistant to metabolic hydrolysis than esters (X=O). Thioesters (X=S) may cause dermatitis.
- In procaine-like analogues, branching (especially at the alpha carbon) will increase duration of action. This effect is not seen in the lidocaine series.
- Increasing the chain length will increase potency but will also increase toxicity.
Aminoalkyl group
- The aminoalkyl group is not necessary for local anaesthetic activity, but it is used to form water-soluble salts (HCl salts).
- Tertiary amines result in more useful agents. The secondary amines appear to be of longer activity, but they are more irritating; primary amines are not very active and cause irritation.
- The tertiary amino group may be diethylamino, piperidine, or pyrrolidino, leading to the products that exhibit essentially the same degree of activity.
- The more hydrophilic morpholino group usually leads to diminished potency.
- Some analogues have no amino group at all, such as benzocaine. They are active but have poor water solubility.
STRUCTURES, SYNTHESIS, AND SAR OF ANILIDES
Agents of this class are more stable to hydrolysis. They are more potent, have a lower frequency of side-effects, and induce less irritation than benzoic acid derivatives.
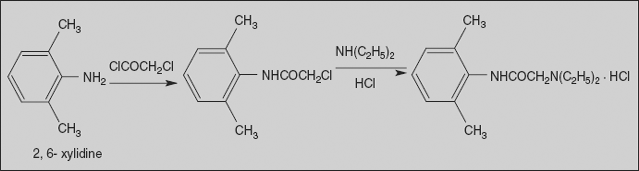
Lidocaine Hydrochloride
Lignocaine, Xylocaine, 2-(Diethylamino)-N-(2,6-dimethylphenyl) acetamide
Synthesis
Lidocaine is synthesised from 2,6-dimethylaniline upon reaction with chloroacetyl chloride, which gives α-chloro-2,6-dimethylacetanilide, and its subsequent reaction with diethylamine affords lidocaine.
It is a widely employed amide-type local anaesthetics extremely resistant to metabolic hydrolysis. In addition to the relative stability of the amide bond, the 2,6-dimethyl substituents provide steric hindrance to attack of the carbonyl. Lidocaine is about twice as potent as procaine. It is also used as an effective cardiac depressant and may be administered intravenously in cardiac surgery and life-threatening arrhythmias.
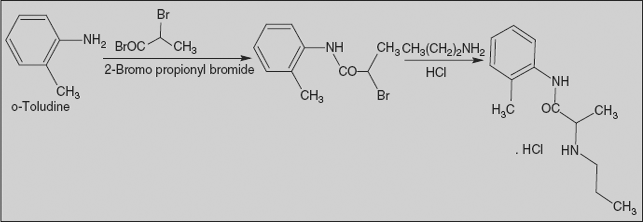
Prilocaine Hydrochloride
N-(2-methylphenyl)-2-(propylamino) propanamide
Synthesis
Prilocaine is synthesised from 2-methylaniline upon reaction with 2-bromopropionyl chloride, followed by reaction with n-propylamine.
Mepivacaine and Bupivacaine Hydrochloride
N-(2,6-Dimethylphenyl)-1-methyl/ butyl-2-piperidine carboxamide
Synthesis
Reaction of 2,6-dimethylaniline with the acid chloride of pyridine carboxylic acid first gives the 2,6-xylidide of α-picolinic acid. The resulting 2,6-xylidide α-picolinic acid is alkylated with the corresponding alkyl halide, followed by reduction with hydrogen in the presence of platinum on carbon catalyst, and affords mepivacaine and bupivacaine.
Etidocaine Hydrochloride
2-(Ethylpropylamino)-2’,6’-butyroxylidide
Synthesis
In the first stage of synthesis, 2,6-dimethylaniline is reacted with α-bromobutyric acid chloride to give the bromoanilide, which on amination with ethyl propyl amine affords etidocaine.
SAR of anilides
General structures of anilides are represented as follows:
Aryl group
- The clinically useful local anaesthetics of this type possess a phenyl group attached to the sp2 carbon atom through a nitrogen bridge.
- Substitution of the phenyl with a methyl group in the 2- or 2- and 6- position enhances activity. In addition, the methyl substituent(s) provide steric hindrance to hydrolysis of the amide bond and enhances the coefficient of distribution.
Substituent X
- X may be carbon, oxygen, or nitrogen. Among them Lidocaine series (X=O) has provided more useful products.
Aminoalkyl group
- The amino function has the capacity for salt formation and is considered the hydrophilic portion of the molecules.
- Tertiary amines (diethylamine, piperidines) are more useful because the primary and secondary amines MISCELLANEOUS
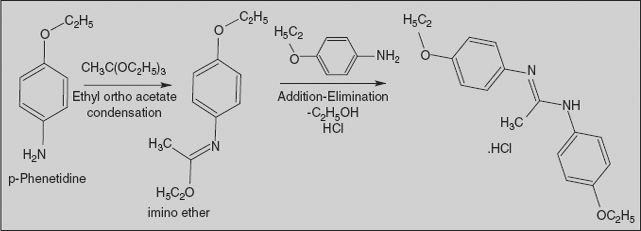
Phenacaine Hydrochloride
N, N’-Bis (4-ethoxyphenyl)-ethanimidamide
Synthesis
Condensation of 4-ethoxyaniline takes place with ethyl orthoacetate to afford the imino ether. Reaction of that intermediate with a second mole of 4-ethoxyaniline results in a net displacement of ethanol, probably by an addition-elimination scheme, and affords phenacaine.
Phenacaine is structurally related to the anilides in that an aromatic ring is attached to an sp2 carbon through a nitrogen bridge. But it lacks the traditional ester or amide function and terminal aliphatic nitrogen.
Diperodon
3-Piperidino-1,2-propanediol dicarbanilate
Synthesis
Alkylation of piperidine with 3-chloro-1,2-propane diol, followed by reaction with two moles of phenyl isocyanate, affords the bis-carbamate diperodon.
Structurally, it is related to the anilides in that an aromatic ring is attached to an sp2 carbon by a nitrogen bridge.
Pramoxine Hydrochloride
4-[3-(4-Butoxyphenoxy)propyl]morpholine
Synthesis
Alkylation of the mono potassium salt of hydroquinone with butyl bromide affords the ether, and alkylation of this with N-(3-chloropropyl)morpholine affords pramoxine.
Structurally, it is unrelated to either ester- or amide-type agents; simple ether linkage fulfils this function that exhibits local anaesthetic activity.
Dyclonine
4’-Butoxy-3-piperidinopropiophenone
Synthesis
4-Hydroxy acetophenone undergoes O-alkylation with butyl bromide, followed by Mannich reaction with formaldehyde and piperidine, and affords dyclonine.
Dibucaine
2-Butoxy-N-(2-(diethylamino)ethyl)cinchoninamide (Cinchocaine)
Synthesis
Acylation of isatin affords N-acetyl isatin, which on treatment with sodium hydroxide affords quinolone derivative. The transformation (Pfitzinger reaction) may be rationalised by assuming the first step to involve the cleavage of lactam bond to afford on intermediate: Aldol condensation of the ketone carbonyl with the amide methyl group leads to the 2-hydroxy cinchoninic acid. Treatment with phosphorous pentachloride serves both to form the acid chloride and to introduce the nuclear halogen. Condensation of the acid chloride with N,N-diethyl ethylenediamine followed by replacement of ring halogen with sodium butoxide affords dibucaine.
Local anaesthetics property of dibucaine (quinoline derivative) was discovered accidentally while research actually aimed for the preparation of antimalarial agents related to quinine. It is the most potent, most toxic, and longest-acting local anaesthetics.
Dimethisoquin Hydrochloride
3-Butyl-1-[2-(dimethylamino)ethoxy] isoquinoline
Synthesis
Condensation of 1-nitropentane with acid aldehyde affords the phthalide via hydroxyl acid. Reduction of the nitro group via catalytic hydrogenation leads to ring opening of the lactone ring to the intermediate amino acid. This cyclizes spontaneously to the isoquinoline derivative. Dehydration by means of strong acid followed by treatment with phosphorous oxychloride converts the oxygen function to the corresponding chloride via the enol form. Displacement of halogen with the sodium salt of 2-(diethylamino) ethanol affords dimethisoquin.
NEWER DRUGS
Newer local anaesthetics were introduced with the goal of reducing local tissue irritation, minimizing systemic cardiac and central nervous system (CNS) toxicity, and achieving a faster onset and longer duration of action.
Ropivacaine
(S)-N-(2,6-Dimethylphenyl)-1-propylpiperidine-2-carboxamide
It was developed after bupivacaine was noted to be associated with cardiac arrest in 0.5–0.75% of cases, particularly in pregnant women. Ropivacaine was found to have less cardiotoxicity than bupivacaine in animal models. Ropivacaine is indicated for local anaesthesia including infiltration, nerve block, epidural, and intrathecal anaesthesia in adults and children over 12 years. It is also indicated for peripheral nerve block and caudal epidural in children aged 1–12 years for surgical pain. It is also sometimes used for infiltration anaesthesia for surgical pain in children.
Levobupivacaine
It is the S-enantiomer of bupivacaine. Compared to bupivacaine, levobupivacaine is associated with less vasodilation and has a longer duration of action. It is approximately 13 per cent less potent (by molarity) than racemic bupivacaine.